Wilson’s disease (WD) is a rare autosomal recessive genetic disorder of copper metabolism that results in accumulation of copper in the liver and subsequently in other organs, mainly the central nervous system. Among the various inherited metabolic disorders, WD carries an importance among all the treating physicians due to potential reversibility and ability to lead a normal life style, if diagnosed early and intervened appropriately. Albeit WD was not reported from India during the first half a century of its description, currently India has become one of hot spots for clustering of WD patients due to various cultural practices and genetic risks. These combined factors of ‘treatment’ and ‘clustering’ of WD patients in India, carries a very important medical intervention factor in the Indian health scenario.
The clinical features of WD are wide and varied with different systems involvement. Because of this varied manifestations diagnostic errors are common and lead to mis-diagnosis. WD can present at any age, but commonly noted at the end of first decade or early second decade of life. The mean age of onset of WD in the largest Indian series was 16 years. About 40% of patients present with hepatic manifestations and another 40% of patients present with neurological manifestations. The remaining subset of patients present with varied systems involvement. The clinical spectrum of WD is wide and variable.
Hepatic involvement and presentation is variable with about 5% of patients present with fulminant hepatic failure with encephalopathy, coagulopathy and Coombs negative haemolytic anemia. All young patients with unexplained chronic liver disease, with or without cirrhosis, should be screened for WD. Hepatocellular carcinoma is rarely associated with Wilson’s disease, but may occur in the setting of cirrhosis and chronic inflammation.
The neurological abnormalities can be classified as: (a) an akinetic- rigid syndrome similar to Parkinson’s disease, (b) pseudosclerosis dominated by tremor, (c) ataxia, and (d) a dystonic syndrome. Subtle signs can appear before the characteristic neurological features, including changes in behaviour, deterioration of school performance, or an inability to carry out activities that need good hand-eye coordination. Hand writing might deteriorate and micrographia could develop. Drooling, dysarthria, seizures may develop. Dystonia of the facial and mandibular muscles produces a characteristic ‘vacuous smile’ with stiff face and open mouth. Along with behavioural changes, other psychiatric manifestations include depression, anxiety and frank psychosis. Patients who first present with neurological or psychiatric signs tend to be older than those with hepatic features alone. Most patients with CNS involvement are believed to have liver disease at the time of presentation but they are often not symptomatic from their liver disease.
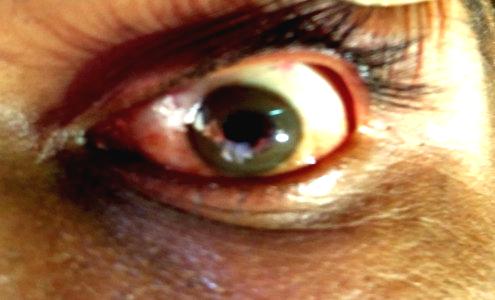
Ophthalmic findings include KF rings and sunflower cataracts. KF rings are caused by the granular deposition of copper on the inner surface of the cornea in Descemet’s membrane. The upper pole is affected first. The rings have golden brown appearance.(Fig.1) Although sometimes visible to the naked eye, slit lamp examination is necessary to confirm the presence or absence of KF rings.
Sunflower cataracts are infrequent and rarely symptomatic.
Osseomuscular manifestations include joint pains, arthritis and non-trauatic fractures. The causes for the metabolic bone disease in WD are manifold and include the following: 1) dietary deficiency of essential minerals and vitamins, 2) renal losses of calcium and phosphorous due to renal tubular acidosis, 3) secondary hypoparathyroidism due to renal dysfunction, 4) hypoparathyroidism probably due to copper deposition in parathyroid glands, and 5) drug related, due to D-penicillamine therapy.
Haematological manifestations include haemolytic anemia due to large amounts of copper released into the blood stream secondary to hepatocellular injury; coagulaopthy secondary to hepatic dysfunction; thrombocytopenia and pancytopenia secondary to hypersplenism or as treatment related adverse effect of penicillamine.
Wilson’s Disease, has come to the forefront among the various metabolic and genetic disorders, primarily due to its potential treatments, which help to lead near normal quality of life, if recognized early and intervened. These treatments can be categorized into
- De-coppering therapies
- Symptomatic Therapies
- Curative therapies
- Asymptomatic Patients
- Special Circumstances
De-coppering therapies:
All symptomatic and all presymptomatic patients require lifelong decoppering. With adequate decoppering neurological disability improves and brain imaging abnormalities reverse.